|

Understanding through Discussion |
|
|
Register | Sign In |
|
QuickSearch
| Thread ▼ Details |
|
Thread Info
|
|
|
| Author | Topic: Discussion of Phylogenetic Methods | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days)  Posts: 1517 From: Michigan Joined:
|
A phylogeny is a hypothesis about the evolutionary history of a group of taxa and since the phylogeny we present is a hypothesis, we want to know how well our hypothesis is supported compared to other hypotheses. Thus, the various phylogenetic methods have been developed to provide researchers with ways to evaluate those hypotheses and determine which hypothesis is the best. In other words, phylogenetic programs don't just build trees but more importantly, they evaluate them so that researchers can present the most well supported hypothesis.
The first thing that needs to be clarified is what is meant by the tree space. The tree space is all possible topologies that a particular combination of taxa could produce. The number of possible bifurcating, rooted trees for a given number of taxa m is given by the formula: (2m - 3)!/[2m-2(m-2)!] So, for just 10 taxa, there are 34,459,425 possible trees in the tree space, which demonstrates that the tree space becomes extremely large with even a small number of taxa and thus makes it all but impossible to evaluate ALL the various trees within the tree space. The assumption is that one of these 34,459,425 trees represents the true evolutionary history of the 10 taxa in question. However, since we can never know for sure which tree is the TRUE tree, what we want to do is propose our best hypothesis as to which tree best represents the true evolutionary history of the taxa. How we do that is by specifying some optimality criteria and then finding the tree that has the highest value for our specified optimality criteria. Again, I don’t think the point that a phylogeny is not (or should not be) presented as the true evolutionary history of a set of taxa cannot be overemphasized. What a phylogeny presents is our best estimate of the evolutionary history of a set of taxa. We evaluate how confident we are in that estimate by the type of optimality criteria used and the statistical support for the topology. For example, if a phylogeny of 20 taxa were presented based on 200 nucleotide characters optimized by parsimony, I would have almost no confidence that the hypothesis was correct; in fact, I would pretty much dismiss it as worthless. However, if those same taxa were evaluated using 5000 nucleotide characters from 4 genes optimized by maximum-likelihood with bootstrap support values >90% on more than 3/4 of the branches, I would be very confident in the hypothesis. It is really about confidence levels, which is why newer phylogenetic methods, such as maximum-likelihood and Bayesian, rely so heavily on statistical models. Genomicus requested that I discuss the Bayesian method since it is probably the least understood method - and the most difficult conceptually. Bayes and Maximum-likelihood (abbr. ML ) are pretty much the standard for phylogenetic analysis these days and often researchers will present the results of both analyses. The other methods are falling out of use, but still have some limited applications where Bayes and ML are not appropriate. However, I think it important to cover these other methods before diving in to Bayesian methods because they explain some key concepts that are needed in order to understand Bayesian concepts. The next post will cover this introductory material and provide the background for the discussion of Bayesian methods. HBD (ABE: Biological Evolution I suppose) Edited by herebedragons, : No reason given.Whoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined:
|
In this post, I want to give brief explanations of phylogenetic methods of neighbor-joining (NJ), parsimony, and maximum-likelihood (ML). I am only going to cover the key principles involved (especially as they will be applicable to the discussion of Bayesian analysis) and the main advantages and disadvantages of the method. If anyone would like more information about a specific method, please ask.
Neighbor-joining Neighbor-joining is the most widely used distance-based method. Distances are calculated based on pairwise comparisons and the tree built with those taxa that are closest genetically being most closely related. Distance measurements can be corrected using different nucleotide substitution models. NJ trees can be evaluated by bootstrapping (bootstrapping will be covered at the end of this post), which allows some statistical power. Advantages:One of the major advantages is that NJ is very fast. Even very large datasets take only a few minutes as opposed to hours or even days with other methods. Another major advantage is that NJ will return a single, best tree (best as far as NJ analysis can determine). Why this is an advantage will become more apparent as the other methods are discussed. Disadvantages:The major disadvantage is that NJ assumes that all lineages evolve at the same rate and that genetic distance is directly proportional to relatedness, which is not necessarily the case. Also, distance analyses have no way to take intermediate steps into consideration but can only consider distances between terminal taxa. Thus NJ has no way to consider the possibilities of reversals and parallel changes in determining relatedness. Parsimony Parsimony, in my opinion, is the most unreliable phylogenetic method. It relies on the assumption that evolution occurs in the fewest steps possible, which is often a faulty assumption. In order to determine the most parsimonious tree, the phylogenetic program must search the tree space and evaluate every tree for the number of steps and select the tree that has the fewest. However, with any more than a few taxa, the tree space becomes so large that it is practically impossible to search every tree within the tree space (PAUP limits a full search to 15 taxa, IIRC). So instead, we use a technique called a heuristic search. A heuristic search is a systematic method of searching the tree space. It begins with a randomly selected tree and through branch swapping, looks for an optimal tree. This optimal tree is a local optimum, since the search does not cover the entire tree space. The process then repeats a number of times, each starting at a different, random place in the tree space. I am not aware of a standard to determine how many times this process should be replicated, but 100 random addition replicates is common. The idea is that this should provide sufficient coverage to find the globally optimized tree. Advantages:The main advantage that I see for parsimony analysis is when evaluating taxa for which DNA data is unavailable, such as fossil species. Parsimony is a close enough approximation in this case (although I think ML and Bayesian analyses can be used with morphological data and so would be a better choice than parsimony). Disadvantages:As already mentioned, parsimony is a weak assumption when it comes to molecular evolution and since there are more rigorous methods available it seems pointless to use parsimony analysis on molecular data. Another major disadvantage is that parsimony analysis often returns multiple most parsimonious reconstructions. My own analysis of 110 taxa and 2100 nucleotide characters returned 2048 most parsimonious reconstructions! How do you choose which tree to present since there is no way to favor one reconstruction over another? What you end up doing is creating a consensus tree, so the tree that is presented is not even an actual tree but an artefactual representation of multiple trees. Another disadvantage is that since the tree space was searched heuristically and not completely, there is a possibility that even though the search found hundreds of most parsimonious reconstructions, there is a tree with few steps somewhere in the tree space. Without evaluating every tree in the tree space, there is no way of knowing the most parsimonious tree has been found. A sufficient number of sequence addition replicates helps to ensure that enough of the tree space has been covered to minimize this problem, but of course, that adds time to the analysis.
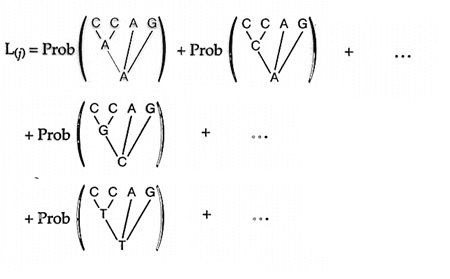
Maximum-likelihood The maximum-likelihood approach asks What is the probability of the observed data given an evolutionary model and a phylogenetic tree? Using an evolutionary model that defines the probability of different nucleotide substitutions (such as what is the probability of an A --> T or a C --> G, etc.), the probability for a site is the sum of the probabilities of every possible reconstruction of ancestral states. The probability for the full tree is the product of the likelihoods at each of the sites. Consider a case of 4 taxa where character 1 is C C A G for taxa 1, 2, 3 & 4 respectively and the topology shown in the figure below where taxa 1 and 2 are sister taxa and there are 2 unobserved ancestral states. In order to determine the likelihood for this tree, we calculate the probability for every possible combination of ancestral character states. The sum of all these probabilities is the probability for site 1. The -Ln ( ) is the likelihood for that site. Now repeat that procedure for every site and sum the -Ln ( ) of all characters and that is the likelihood for the full tree.
As you can imagine, these calculations can be very, very computationally demanding and they need to be done for every possible topology. Like parsimony, maximum-likelihood uses a heuristic method to search the tree space. Advantages:Maximum-likelihood is a very rigorous method that considers possible ancestral states and can be bootstrapped for very good statistical confidence. Although it is possible that there will be more than 1 topology with the best likelihood value, typically ML analysis will return a single, best tree. This is an advantage because the maximum-likelihood tree can be presented as the favored hypothesis. Disadvantages:Extremely time consuming and requires tremendous computational resources. For my project of 110 taxa, I figured it was going to take about 7 days to run 50 replicates in a heuristic search plus another 7 - 14 days for 50 bootstrap replicates. Newer ML programs have been developed that are considerably faster. RaxML and PHyML are a couple examples. I was able to complete my analysis in about 4 hours (as opposed to 14+ days for the heuristic search) for 100 bootstrap replicates using PHyML. Honestly though, I am not really all that confident in the results. Not many of the branches had good support where by other methods most of them were quite well supported. Bootstrapping Bootstrapping is a widely used method that can provide a measure of support for the branches of a phylogenetic tree. In order to create a bootstrap replicate, a new dataset (of the same size as the original) is created by randomly choosing characters (with replacement) from the original dataset. For example, a bootstrap replicate made from a dataset with 10 characters (numbered 1 - 10) might include characters 1, 3, 4, 4, 7, 8, 8, 8, 9, 10. So, characters 2, 5 & 6 are not represented in the replicate while 4 & 8 are represented multiple times. The new dataset is then analyzed the same way the original dataset was. This process is repeated the specified number of times. A consensus of the trees generated from the replicates is created and the bootstrap support value for a branch is the percentage of times that particular branch appears in the set of replicate trees. The reasoning behind this process is that if a branch is well supported there should be a significant number of characters that support that topology and by selecting characters at random, the strength of the phylogenetic signal should be detectable. The downside to this process is that not only is the tree that will be presented a consensus, but it is also created from artificial data.------------ This post should provide the background to the principles and theories that led to the development of Bayesian analysis. I realize that there is a lot of information here but I tried to be as brief as I felt I could be. Hopefully it is all relatively clear but I expect that in my attempt at brevity, I left some explanations vague or unclear. I would appreciate questions, comments or discussion regarding anything related to this topic. I would also expect that my comments about parsimony may be controversial and might generate some discussion which would also be welcomed. I will come back to discuss Bayesian analysis as soon as possible. HBDWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Admin Director  Posts: 13014 From: EvC Forum Joined: Member Rating: 1.9 |
I'd like to make sure this can be easily understood by rank laypeople.
herebedragons writes: A phylogeny is a hypothesis about the evolutionary history of a group of taxa... About "phylogeny," is this the way biologists actually use the term? I'm asking because when I look "phylogeny" up in the dictionary it seems like the above could be more easily understood if it were phrased like this: "A phylogenetic tree is a specific hypothesis about the evolutionary history of a group of taxa..."
Thus, the various phylogenetic methods have been developed to provide researchers with ways to evaluate those hypotheses and determine which hypothesis is the best. A natural objection might be that selecting the best among a bunch of poor hypotheses is not of much value. What tells us that the better hypotheses have a fair chance of being true? You go on to describe some evaluation criteria like optimality, but giving a name to a criteria in this case explains little.
For example, if a phylogeny of 20 taxa were presented based on 200 nucleotide characters optimized by parsimony,.. "Nucleotide characters" means groups of three nucleotides that program for amino acids? Or do just mean individual nucleotides?
However, if those same taxa were evaluated using 5000 nucleotide characters from 4 genes optimized by maximum-likelihood with bootstrap support values >90%... You might be descending into jargon here. I don't have time now to tackle your next post.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined: |
About "phylogeny," is this the way biologists actually use the term? A phylogeny is the evolutionary history of a group of organisms and the result of a phylogenetic analysis. A phylogenetic tree is a graphic representation of a phylogeny. The phylogeny itself is the hypothesis about the evolutionary history of a group of taxa. I could try to clarify that better.
What tells us that the better hypotheses have a fair chance of being true? You go on to describe some evaluation criteria like optimality, but giving a name to a criteria in this case explains little. I explain the optimality criteria more in depth in the next post.
"Nucleotide characters" means groups of three nucleotides that program for amino acids? Or do just mean individual nucleotides? This issue would need to be resolved during alignment. Once the alignment is done the phylogenetic analysis treats each individual nucleotide as a separate character. The alignment is critical to any phylogenetic analysis but I wasn't sure there would be interest in a prolonged discussion about alignment, so I was trying to gloss over it.
You might be descending into jargon here. I could probably delete that whole paragraph as it was just meant to be a quick example about how confidence affects our conclusions. I could save that for later.
I don't have time now to tackle your next post. I will wait to make any corrections until I get your comments on that post. HBDWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Admin Director Posts: 13014 From: EvC Forum Joined: Member Rating: 1.9
|
Thread copied here from the Discussion of Phylogenetic Methods thread in the Proposed New Topics forum.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Genomicus Member (Idle past 1960 days)  Posts: 852 Joined:
|
Thanks, herebedragons! Will be commenting on this in a bit in more depth. You've broken this down in an easier-to-understand manner than what you'd find in a lot of bioinformatics textbooks.
I'd definitely encourage the broader EvC community to weigh in on this topic with their own questions or comments of relevance to phylogenetics.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Dr Adequate Member (Idle past 303 days)  Posts: 16113 Joined:
|
An excellent thread, keep going.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Tanypteryx Member  Posts: 4407 From: Oregon, USA Joined: Member Rating: 5.5
|
HBD, thanks for starting this thread.
I have several questions, but I am not sure where in the discussion you would want to answer them. If you answer them, where ever in this discussion you think is right is fine with me. If you were going to try to create a phylogeny for a family that contains 11 species in 8 genera, how would you decide which regions of the genome to sequence? Do you want the regions to be non-coding or regulatory? How many regions would you want to sequence for the family phylogeny? How many regions to determine the amount of genetic diversity, within and between populations a single species? How many regions to try and construct a genetic clock? Could the same data collected be used for all these determinations? Cheers.What if Eleanor Roosevelt had wings? -- Monty Python One important characteristic of a theory is that is has survived repeated attempts to falsify it. Contrary to your understanding, all available evidence confirms it. --Subbie If evolution is shown to be false, it will be at the hands of things that are true, not made up. --percy
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Taq Member  Posts: 10024 Joined: Member Rating: 5.3 |
"Nucleotide characters" means groups of three nucleotides that program for amino acids? Or do just mean individual nucleotides? It might be helpful to mention that non-coding DNA can also be used for constructing phylogenies. Obviously, non-coding DNA is never translated into a peptide, so there are no 3 base codons in non-coding DNA. However, I would be interested to see if the Maximum Likelihood method takes advantage of synonymous vs. non-synonymous mutations. From the description above: "Using an evolutionary model that defines the probability of different nucleotide substitutions (such as what is the probability of an A --> T or a C --> G, etc.), the probability for a site is the sum of the probabilities of every possible reconstruction of ancestral states." Substitutions in the first two bases of a codon are much more likely to cause a detrimental mutation and be selected against. Mutations in the 3rd base may not change the amino acid sequence at all. The question I have for HBD is if synonymous and non-synonymous mutations are weighted differently in this method.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined:
|
Good questions Tanypteryx. The simple answer is... it depends.
Essentially they are questions about alignment, as is Taq's in the next post. A good alignment (including choosing appropriate genes regions) is critical to a good phylogenetic study. I will plan to spend some time later on alignments. But for now I will give some brief answers to your questions.
How many regions would you want to sequence for the family phylogeny? Four seems to be the minimum number of gene regions for an acceptable phylogenetic study; I have seen 8, but I can't think of any studies that have used more than that, so I would say 4 to 8 genes would be good.
If you were going to try to create a phylogeny for a family that contains 11 species in 8 genera, how would you decide which regions of the genome to sequence? First, I would find out what other researchers are using for closely related species; this would be a good starting point. There are also some genes that are commonly used that would probably work for most Eukaryotes. - Ribosomal RNA (rRNA) includes the 5.8S, 28S and 18S subunits as well internal spacers (ITS1 and ITS4). The subunits are transcribed into RNA but not translated into proteins. The internal spacers are also transcribed but snipped out before the subunits are assembled. - Transcription Elongation Factor 1-alpha (EF-1a), RNA polymerase II subunits RPB1 and RPB2, Beta-tubulin and Histone H3 are some common nuclear genes used. - Mitochondrial genes cytochrome c oxidase (cyt c) and rRNA 16S
Do you want the regions to be non-coding or regulatory? We should frame this question in a different way... Do we want the regions to be highly conserved or highly variable? (Non-coding regions tend to be highly variable and coding regions tend to be conserved) For organisms that are very closely related, highly conserved regions will have little informative information, or in other words, too many of the subjects will have identical sequences. Conversely, regions that are highly variable will be virtually impossible to align in distantly related species. So, the type of region you choose depends on the species being studied. Often genes are combinations of both types, coding and non-coding (exons and introns respectively). The coding regions are able to align well (since they are conserved) and the non-coding regions provide the phylogenetic informative characters. The figure below shows the ITS region, a widely used gene region. Using the primers ITS1 and ITS4 produces a fragment that contains part of the 18S and 28S subunits (coding), the entire 5.8S subunit (coding) and the two non-coding spacers between them.
How many regions to try and construct a genetic clock? I am not really sure about this. I know there needs to be a way to calibrate the clock, meaning there needs to be a known time frame and a known number of substitutions. I also know that different genes evolve at different rates, so a molecular clock may only consider one gene. Other than that, I am not very familiar with the molecular clock techniques.
How many regions to determine the amount of genetic diversity, within and between populations a single species? Genetic diversity studies are somewhat different than phylogenetics and I am not that familiar it yet. However, I am expecting to start a diversity study using microsatellites later this spring. So maybe more information later. HBDWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined: |
Substitutions in the first two bases of a codon are much more likely to cause a detrimental mutation and be selected against. Mutations in the 3rd base may not change the amino acid sequence at all. The question I have for HBD is if synonymous and non-synonymous mutations are weighted differently in this method. I am not aware of an alignment program that weighs the third codon differently. However, what they do is convert the codons to amino acids and then align the amino acids. A matrix is used to weight the amino acid substitutions where synonymous substitutions would have high scores and non-synonymous substitutions would have low scores (the alignment algorithm would try to maximize the alignment score). Below is an example of such a matrix (BLOSUM62).
HBDWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Taq Member Posts: 10024 Joined: Member Rating: 5.3 |
I am not aware of an alignment program that weighs the third codon differently. However, what they do is convert the codons to amino acids and then align the amino acids. A matrix is used to weight the amino acid substitutions where synonymous substitutions would have high scores and non-synonymous substitutions would have low scores (the alignment algorithm would try to maximize the alignment score). Below is an example of such a matrix (BLOSUM62). Perhaps I am being overly critical or getting the terminology wrong, but in my understanding a synonymous mutation is one that results in the same amino acid. What your chart seems to be using is groupings based on charge and hydrophobicity. A substitution that replaces a polar amino acid with a hydrophobic amino acid is weighted less than a substitution with another polar amino acid of similar charge.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined:
|
Perhaps I am being overly critical or getting the terminology wrong, but in my understanding a synonymous mutation is one that results in the same amino acid. No, you are exactly right. I just didn't do a good enough job explaining how it is handled. Maximum-likelihood calculations do not take into account codon position, but it is not necessary to do so; there is a probability associated with an A mutating into a G regardless of its position. Whether the mutation results in a viable or a non-functioning protein will depend on what position the mutation occurred in but for the purposes of determining phylogenetic relationships, the important thing is that the mutation occurred and that it can be aligned properly. The important part of the process is aligning the sequences so that the codons do line up and third position codons are compared to third position codons (as well as the other 2 positions, but you specifically asked about the third position). Consider the following protein coding sequences that we want to align and infer their relationship: 1 - CGT GGG AAA2 - CGG GGA AAA If you tried to align them like this, the algorithm might insert a gap so that the nucleotides line up better. It could look like this: 1 - CGT GGG AAA2 - CG- GGG AAA A which would be an inappropriate alignment. Instead, the codons are kept together and converted into the appropriate amino acids and then aligned. So, after converting to amino acids the alignment looks like this 1 - Arg - Gly - Lys2 - Arg - Gly - Lys The chart I presented describes how the alignment algorithm determines whether to align 2 non-synonymous substitutions or insert a gap. (Synonymous mutations would be substituting the same amino acid eg.: Trp --> Trp = 11). And you are exactly right, a polar amino acid is more likely to align with another polar amino acid than it is to a hydrophobic amino acid. Once the alignment is done, it is converted back into codons and then the phylogenetic algorithm evaluates the sequences base by base, treating each base as an individual character. So our example becomes
The maximum-likelihood is calculated for character 1 (likelihood of C --> C); then character 2 (G --> G); character 3 (T --> G); etc... Since each character is evaluated independently, its position is irrelevant. I hope that better explains how coding sequences are handled in phylogenetics. It looks like I need to plan on explaining the whole alignment process better since it is such an important part of the whole process and seemingly not well understood. HBD Edited by herebedragons, : typoWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Genomicus Member (Idle past 1960 days) Posts: 852 Joined:
|
How many regions to try and construct a genetic clock? Generally speaking, the more proteins used in a molecular clock analysis, the better. For your example -- a family that contains 11 species in 8 genera -- you'd want to avoid highly conserved proteins (e.g., serum albumin, cytochrome c), as there probably wouldn't be enough substitutions in these proteins across these closely related taxa to create a reliable molecular clock. For more accurate results, you would also want divergence times of at least a couple of these species as determined by paleontology. In this way, you can calibrate the molecular clock for this family of organisms. Edited by Genomicus, : No reason given.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
herebedragons Member (Idle past 876 days) Posts: 1517 From: Michigan Joined:
|
Maximum-likelihood evaluates trees based on the probability that evolution would produce the observed data. Bayesian inference evaluates trees based on their posterior probability, which is the probability that the tree is true given the data, based on an evolutionary model and taking into consideration the prior probability. To get a feel for how the Bayesian method works let’s look at an example.
You are given a bag of coins and told that 1/2 of the coins are fair (50% heads) and 1/2 are biased (75% heads). One coin is selected from the bag at random and you wish to consider the two alternative hypotheses that either the coin is fair or it is biased. The coin is flipped 10 times and each time it falls on heads. Now we can apply likelihood to determine whether our data (10 heads) supports one hypothesis over the other. The likelihood is the probability that the data would have arisen from a given hypothesis, ie. the coin is fair. With 10 heads the likelihood is the product of the probabilities of each toss or 0.510 = 0.00098. This does not mean there is a 0.1% chance the coin is fair, it means there is a 0.1% chance of this specific outcome for a fair coin. That is, there is a 0.001 probability of the data given the specific hypothesis. The probability of the data given the hypothesis that the coin is biased is 0.7510 = 0.0563, which is still a really low number, which tells us that even under the biased hypothesis this outcome is highly unlikely. What counts however, is the comparison of the likelihoods of the competing hypotheses. The ratio of likelihoods is 0.0563/0.00098 or about 57 times as probable under the biased hypothesis as it is under the fair hypothesis. This likelihood ratio is usually expressed as a natural logarithm and is a measure of support for one hypothesis over another. In this example, ln(57) = 4.05, which is well above the commonly used threshold of 2.0 (which approximates the traditional P < 0.05 confidence level). We would conclude that the data strongly supports the conclusion that the coin is biased. With likelihood we are able to deduce that the hypothesis that the coin is biased is more likely to produce the observed data than the hypothesis that the coin is fair, but we did not calculate the actual probability that the coin was biased. Bayesian theorem states that the probability of a hypothesis given some data, is equal to the probability of the data given the hypothesis (the likelihood) times the prior probability divided by the total probability of the data (summed over all hypotheses).
Where:
Now we can apply this theorem to our coin flipping example to determine what the probability is that we have selected a biased coin given the data of flipping 10 heads in a row. Using the same model as before, we can determine The denominator is a bit more difficult to determine. Here, there are only 2 possible hypotheses; that the coin is fair or that it is biased. There is a 0.5 chance it is fair, and if it is, the probability of getting 10 heads is 0.510. There is also a 0.5 chance the coin is biased, and if it is, the probability of getting 10 heads is 0.7510. Summing these together is the total probability of the data 0.5 x (0.510 + 0.7510) = 0.0286. This means there is a 2.9% chance of selecting a coin at random and then obtaining 10 heads in 10 tosses. Combining these numbers gives us 0.0281 / 0.0286 = 0.98 or 98%. This means that given the data, the priors and the model, there is a 98% chance that the coin is biased and only a 2% chance that the coin is fair. By accounting for the observation of flipping 10 heads, the probability that a biased coin was selected went from 0.5 to 0.98. The notable aspect of the Bayesian approach is that the starting information matters. If the original sample from which the coin was selected only contain 1% biased coins, the posterior probability would only be 0.37
So in this case, even after observing 10 heads, it would still be better to bet against the coin being biased. Now we need to apply these principles to phylogenetics. The data corresponds to a character state matrix and the hypotheses correspond to the alternative tree topologies. Thus Bayes’ theorem takes the form:
The prior probability of a particular tree is the probability that among all possible tree topologies it is the correct one. If we believed that all trees were equally likely, then we could assign a flat prior, where the prior probability of a tree equals one divided by the number of trees. The probability of the data given the tree is calculated as the maximum likelihood as described earlier. The difficulty comes in calculating the probability of the data which requires a summation over all possible tree topologies. Maximum-likelihood and parsimony can assign a score to a tree in isolation but a Bayesian posterior probability cannot be assigned to a single tree without taking into account all possible trees. Instead of calculating the prior probability for each individual tree, Bayesian analysis uses a system known as a Marcov chain Monte Carlo to compare the relative posterior probabilities of different trees, which is a concept I will discuss in the next post. The probability of the data, Pr(Data) (the denominator of the Bayesian equation) will be the same across all possible trees and will therefore cancel out when the ratio is calculated. I am going to have to stop here and discuss the next part of the process later. I apologize that this post was so theoretical and I thought about not bringing it up, but I felt it is actually an important part of the Bayesian analysis and without this background understanding, it may not make as much sense as to why the method has developed the way it has. I am hoping that it becomes more clear how the Bayesian theorem influenced the development of the methodology and why the process works the way it does. HBD Reference: Most of this post was based on material presented in (a highly recommended book, by the way): Baum, D. A., Smith, S. D. (2013). Tree thinking: An introduction to phylogenetic biology. Roberts and Company Publishers, Greenwood Village, CO., USA. Edited by herebedragons, : spellingWhoever calls me ignorant shares my own opinion. Sorrowfully and tacitly I recognize my ignorance, when I consider how much I lack of what my mind in its craving for knowledge is sighing for... I console myself with the consideration that this belongs to our common nature. - Francesco Petrarca "Nothing is easier than to persuade people who want to be persuaded and already believe." - another Petrarca gem. Ignorance is a most formidable opponent rivaled only by arrogance; but when the two join forces, one is all but invincible.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Do Nothing Button
Copyright 2001-2023 by EvC Forum, All Rights Reserved
![]() ™ Version 4.2
™ Version 4.2
Innovative software from Qwixotic © 2024